SynGenome
Going beyond the observable evolutionary universe
DNA is the fundamental layer of biological information in all living organisms. Researchers have made tremendous progress in developing technologies for reading, writing, and editing DNA, but composing new DNA sequences remains a formidable challenge. Advanced AI models, trained on genomic data from millions of organisms, now enable us to compose new genomic sequences beyond what has been generated by natural evolution.
SynGenome is a first-of-its-kind database consisting of synthetic DNA sequences generated by Evo, a genomic language model. Evo is similar to models of human language that learn to predict the next word in a sentence. Given a DNA sequence prompt, Evo will generate a DNA sequence response that continues the genomic sequence. Essentially, Evo enables “autocomplete” for the genome.
In the genomes of prokaryotes and phage, genes with related functions frequently appear directly next to each other along the DNA sequence. As a result, prompting Evo with a sequence encoding a function of interest instructs the model to generate functionally related genes. This enables function-guided generative design by prompt engineering a genomic language model.
SynGenome is organized according to the known functions, domains, and species of the prompt sequences. The corresponding response sequences are likely enriched for genes with related functions or domains, but they could contain many other interesting genes as well. The generated sequences in SynGenome may be very different from anything found in nature while still performing useful biological functions, opening up a new universe of biological discovery.
FAQs
How was SynGenome made?
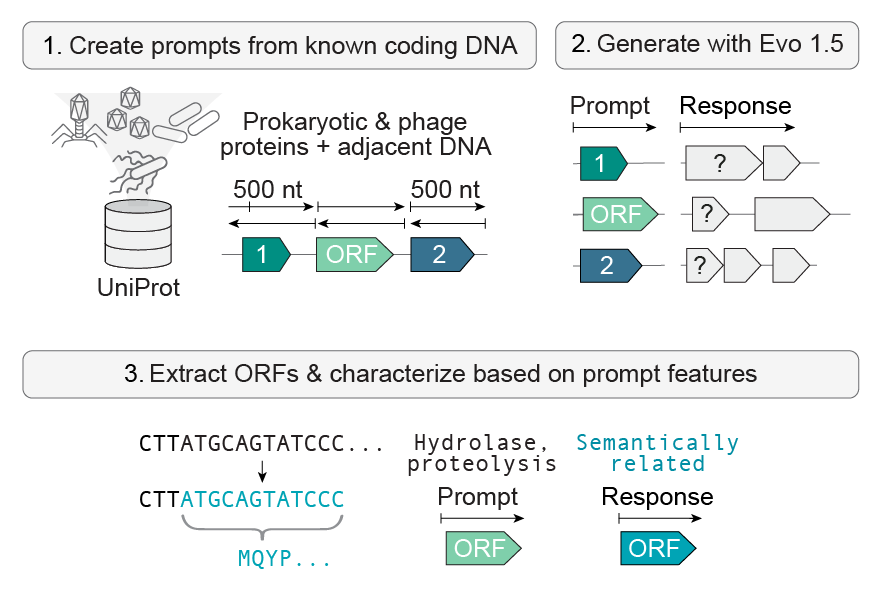
SynGenome contains more than 100 billion DNA base pairs generated by Evo, a DNA language model trained on millions of prokaryotic genomes. Evo is trained to predict the next base pair in a DNA sequence, which enables Evo to generate a response sequence that follows a DNA prompt. To prompt Evo, we used DNA sequences encoding or directly adjacent to known protein-coding genes from prokaryotic organisms and bacteriophages (as compiled by the UniProt database). While the prompt sequences come from natural genomes, the responses can be very different from any DNA sequence found in nature.
What do the “function,” “domain,” and “species” labels mean?
SynGenome entries are organized by the labels associated with the prompt sequence as provided by UniProt. Functional annotations take the form of Gene Ontology (GO) terms. Protein domain annotations are based on InterPro identifiers. Finally, the species labels correspond to the organism from which the prompt sequence was derived.
How should I use SynGenome?
Many important biological systems, with applications in genome editing or DNA synthesis, have been found by searching through genomic databases in a process referred to as “genome mining.” The sequences in SynGenome could be mined similarly. But rather than mining natural sequences, these sequences are generated based on their relationship with known genes in the prompt and can be unlike natural sequences, which is why we refer to this process as “semantic design.” Because the synthetic sequences are likely enriched for functions that are related to the prompt sequence, these sequences could form the basis of a functional screening library.
Have any sequences in SynGenome been experimentally validated?
Using the same strategy for generating sequences in SynGenome, we have had high experimental success rates in low-throughput experiments in which we test only tens of variants for the desired function encoded by a genomic prompt. This includes the design of proteins with anti-CRISPR activity, as well as the design of both bacterial toxins and their conjugate antitoxins. See our paper for more details.
Methodology
Prompt construction
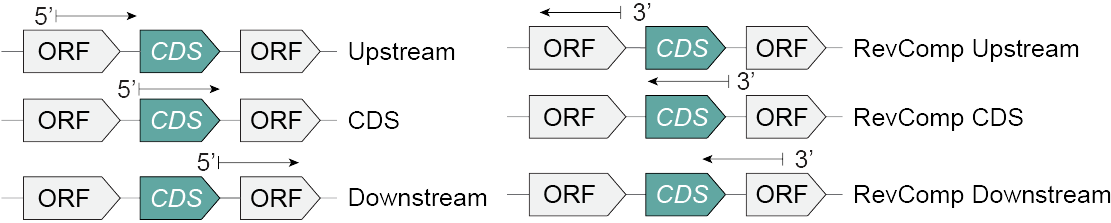
Each prompt is associated with the coding sequence (CDS) of a UniProt entry. We constructed six prompts denoted in the raw data with different labels. A diagram illustrating these prompts and a table describing the prompt type labels are below.
| Prompt type | Description |
|---|---|
| Upstream | The 500 base pair (bp) sequence at the 5' end of the coding sequence (CDS). |
| CDS | The 500 bp sequence beginning with the CDS. If the CDS sequence is shorter than 500 bp, the CDS sequence is rounded down to the nearest 100 bp. |
| Downstream | The 500 bp sequence at the 3' end on the CDS. |
| RevComp Upstream | The reverse complement of the 500 base pair sequence at the 5' end of the CDS. |
| RevComp | The reverse complement of the 500 base pair sequence that ends with the 5' end of the CDS. If the reverse complement of the CDS sequence is shorter than 500 bp, the CDS sequence is rounded down to the nearest 100 bp. |
| RevComp Downstream | The reverse complement of the 500 base pair sequence at the 3' end of the CDS. |
Model and sampling algorithm
We generate sequences with the Evo 1.5 model without any additional finetuning or post-training. We use a standard autoregressive decoding algorithm to sample new sequences with a temperature of 0.7, top-k of 4, and top-p of 1. Code for generating sequences with Evo can be found here. For each prompt type described above, we sampled 2 sequences.
Post-processing
After sampling, we ran NCBI dustmasker to remove highly repetitive sequences, for example, long stretches of sequence containing only a single base pair. While many repetitive sequences were eliminated, we chose not to filter out all repetitive elements given that some prokaryotic genomes do naturally contain biologically meaningful repeats.
Raw data format
The raw SynGenome data is provided in CSV format. Below are the column names and a brief description of the field. Fields corresponding to sequences generated by Evo are in bold.
| Field | Description |
|---|---|
| UUID | Unique identifier for each entry in the dataset |
| Prompt | Input text used to generate sequences |
| Generated_Seq | DNA sequence output generated from the prompt |
| Score | Evo log-likelihood of the sequence |
| File_Derivation | Source file |
| UniProt_CID | ID of the protein |
| Type | Classification of the prompt (e.g., Upstream, Downstream, CDS) |
| Entry | UniProt accession |
| Organism | Name of the biological organism |
| Gene Ontology IDs | Unique identifiers from Gene Ontology database (e.g., GO:0016020) |
| Gene Ontology (biological process) | GO terms describing biological processes |
| Gene Ontology (cellular component) | GO terms describing cellular locations |
| Gene Ontology (GO) | All Gene Ontology annotations |
| Gene Ontology (molecular function) | GO terms describing molecular activities |
| Protein families | Classification of protein family membership |
| CDD | CDD domain annotations |
| DisProt | DisProt database annotations |
| Gene3D | Gene3D database annotations |
| HAMAP | HAMAP database annotations |
| InterPro | InterPro domain annotations |
| NCBIfam | NCBI protein family annotations |
| PANTHER | PANTHER database annotations |
| Pfam | Pfam database annotations |
| PIRSF | PIRSF database classifications |
| PRINTS | PRINTS database annotations |
| PROSITE | PROSITE database annotations |
| SFLD | SFLD database annotations |
| SMART | SMART database annotations |
| SUPFAM | SUPFAM database annotations |
| Domains Compiled | Compilation of domain annotations from various sources |
| Gene Names | Official or common names of genes |
| Protein names | Names or descriptions of proteins |
| Generation_Proteins | Generated protein sequences contained in the DNA response |
Domain Association Network
The SynGenome Domain Association Network visualizes the relationships between protein families that co-occur in prompt-response pairs within SynGenome. When Evo generates DNA sequences in response to prompts, it often produces genes encoding protein families that are functionally related to those found in the prompt sequence. This network captures such associations by drawing edges between Pfam families found in prompt sequences (source nodes) to those in generated responses (target nodes). The weight of each edge corresponds to how frequently a given domain association appears across all prompt-response pairs in SynGenome. To support higher-level functional association, both nodes and edges are colored based on their Pfam Clan membership. For efficient visualization and exploration, the network only displays the top 10,000 domain associations by frequency, with a maximum of five strongest edges displayed per node. Users can search for protein functions or families of interest by keyword, Pfam ID, or Pfam Clan, and interactively explore the network by clicking on nodes to view their connected neighbors. We hope this network can be a starting point to help users better explore the generated diversity in SynGenome, find relevant prompts to apply semantic design to generate their own proteins of interest, and better understand the functional relationships captured by Evo.
Limitations
When using SynGenome, please keep in mind a few important details. All of the generated DNA sequences are synthetic sequences and any downstream functional claims regarding these sequences should be based on additional experimental data. To achieve high experimental success rates, some bioinformatic filtering related to your function of interest is recommended. The “function,” “domain,” and “species” labels in SynGenome are based on the UniProt annotations of the prompt sequence. While the response sequences may be enriched for the same GO terms or InterPro domains as in the prompt, there could also be a diversity of biological structures and functions contained in the response. Generating sequences with a language model is prone to highly repetitive generations; while many trivially repetitive regions have been filtered out, some repetitive sequences, especially those with more complex motifs, have been retained.
Safety
Evo is trained on datasets that explicitly exclude eukaryotic viruses, and semantic design is applied only to prokaryotic genomes, focusing the approach on functions that generally pose low risk to humans. We have also removed generations associated with select agents or regulated toxins, and prompts are functionally annotated so that sensitive categories can be identified. Finally, in practice, semantic design still requires extensive downstream filtering, screening, and optimization, and the SynGenome sequences should be treated as starting points that undergo additional screening review, as opposed to immediately testable constructs.
Acknowledgements
SynGenome makes use of and is inspired by other biological sequence databases such as the UniProt database of protein sequences, the Gene Ontology database of gene annotations, and the InterPro database of protein domain annotations. Evo is trained on data from the Genome Taxonomy Database (GTDB) and the IMG/PR and IMG/VR databases from the Joint Genome Institute. We extend our gratitude to the developers and maintainers of these resources.
Download
The full database is available for download at Hugging Face datasets: https://huggingface.co/datasets/evo-design/syngenome-uniprot.
For downloads specific to a given GO term, InterPro domain, species, or UniProt ID, please use the SynGenome browse/search functionality and click the “Download prompts and generations” button on a given entry page.
We also provide ~3.7 million protein structures predicted by ESMFold of predicted protein coding sequences in SynGenome. We preferentially folded sequences based on the presence of protein family domain hits. These structures are also available at Hugging Face datasets: https://huggingface.co/datasets/evo-design/syngenome-protein-structures
License and citation
SynGenome is freely available under an MIT license. If this database or any of its contents prove useful for your research, please cite Merchant et al. (2025).
@article {merchant2025semantic, author = {Merchant, Aditi T and King, Samuel H and Nguyen, Eric and Hie, Brian L}, title = {Semantic design of functional de novo genes from a genomic language model}, year = {2025}, doi = {10.1038/s41586-025-09749-7}, URL = {https://www.nature.com/articles/s41586-025-09749-7}, journal = {Nature} }